SIMULATION & MODELING
Simulation & Modeling
MFA, KFKI Campus, Building 26.
Contact: Péter Süle, sule @ mfa.kfki.hu
Personal: Péter Süle, Márton Szendrő
This group is concerned with atomistic computer simulations of various 2D materials. Of primary interest is the development of methologies for modelling supported graphene. Correlation between theory and experiment is of major interest as is the comparison of results obtained using different methodologies. Typical approaches in current or recent use include classical molecular dynamics as well as first principles calculations (DFT). We employ predictive modelling techniques in close collaboration with experiments to calculate, understand and predict a wide range of materials properties.
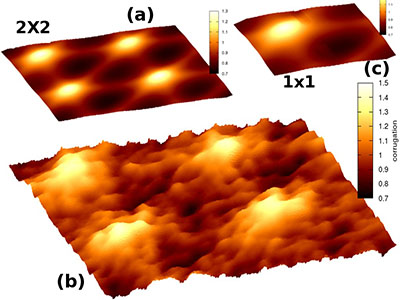
Superlattice of graphene on Cu(111)
Graphene research:
• moiré superlattices
• modelling of graphene/support interfaces
• band structure, LDOS, charge distribution of 2D systems
• gr/Cu(111), gr/Au(111), gr/Ru(0001), MoS2/Au(111)
• new force fields for supported graphene
Main features:
• massively parallel large scale computer simulations (MPI)
• classical molecular dynamics (LAMMPS)
• ab initio Density Functional Theory (SIESTA, CP2K, Quantum Espresso)
• development of new force fields for interfaces
• van der Waals DFT approaches (Grimme, nonlocal DF2)
• geometry relaxation, simulated annealing, computer simulations
• projection of results: OVITO, Gnuplot, GIMP etc.